 中文网站
中文网站
En la madrugada del 29 de diciembre, el NEJM publicó en línea un nuevo estudio clínico de fase III sobre el nuevo coronavirus chino VV116. Los resultados mostraron que VV116 no era peor que Paxlovid (nematovir/ritonavir) en términos de duración de la recuperación clínica y presentaba menos efectos adversos.

Fuente de la imagen: NEJM
Tiempo medio de recuperación: 4 días; tasa de eventos adversos: 67,4 %.
VV116 es un fármaco nucleósido oral contra el nuevo coronavirus (SARS-CoV-2) desarrollado en colaboración con Junsit y Wang Shan Wang Shui, y es un inhibidor de RdRp junto con el remdesivir de Gilead, el molnupiravir de Merck Sharp & Dohme y la azelvudina de Real Biologics.
En 2021, se completó en Uzbekistán un ensayo clínico de fase II de VV116. Los resultados del estudio demostraron que el grupo tratado con VV116 experimentó una mejoría en los síntomas clínicos y redujo significativamente el riesgo de progresión a la forma crítica y de muerte en comparación con el grupo de control. Gracias a los resultados positivos de este ensayo, VV116 fue aprobado en Uzbekistán para el tratamiento de pacientes con COVID-19 de moderada a grave, convirtiéndose en el primer fármaco coronario oral aprobado para su comercialización en el extranjero en China [1].
Este ensayo clínico de fase III[2] (NCT05341609), dirigido por el Prof. Zhao Ren del Hospital Ruijin de Shanghái, el Prof. Gaoyuan del Hospital Renji de Shanghái y el Académico Ning Guang del Hospital Ruijin de Shanghái, se completó durante el brote causado por la variante Omicron (B.1.1.529) de marzo a mayo en Shanghái, con el objetivo de evaluar la eficacia y seguridad de VV116 frente a Paxlovid para el tratamiento temprano de pacientes con COVID-19 leve a moderado. El objetivo fue evaluar la eficacia y seguridad de VV116 frente a Paxlovid para el tratamiento temprano de pacientes con COVID-19 leve a moderado.

Fuente de la imagen: Referencia 2
Entre el 4 de abril y el 2 de mayo de 2022 se llevó a cabo un ensayo multicéntrico, aleatorizado, controlado y con enmascaramiento para el observador, con 822 pacientes adultos con Covid-19 de alto riesgo de progresión y con síntomas leves a moderados, para evaluar la elegibilidad de los participantes de siete hospitales en Shanghái, China. Finalmente, 771 participantes recibieron VV116 (384, 600 mg cada 12 horas el día 1 y 300 mg cada 12 horas los días 2 a 5) o Paxovid (387, 300 mg de nimatuvir + 100 mg de ritonavir cada 12 horas durante 5 días) como medicación oral.
Los resultados de este estudio clínico demostraron que el tratamiento temprano con VV116 para la COVID-19 leve a moderada cumplió con el criterio de valoración principal (tiempo hasta la recuperación clínica sostenida) previsto por el protocolo clínico: el tiempo medio hasta la recuperación clínica fue de 4 días en el grupo de VV116 y de 5 días en el grupo de Paxlovid (cociente de riesgos, 1,17; IC del 95 %, 1,02 a 1,36; límite inferior >0,8).
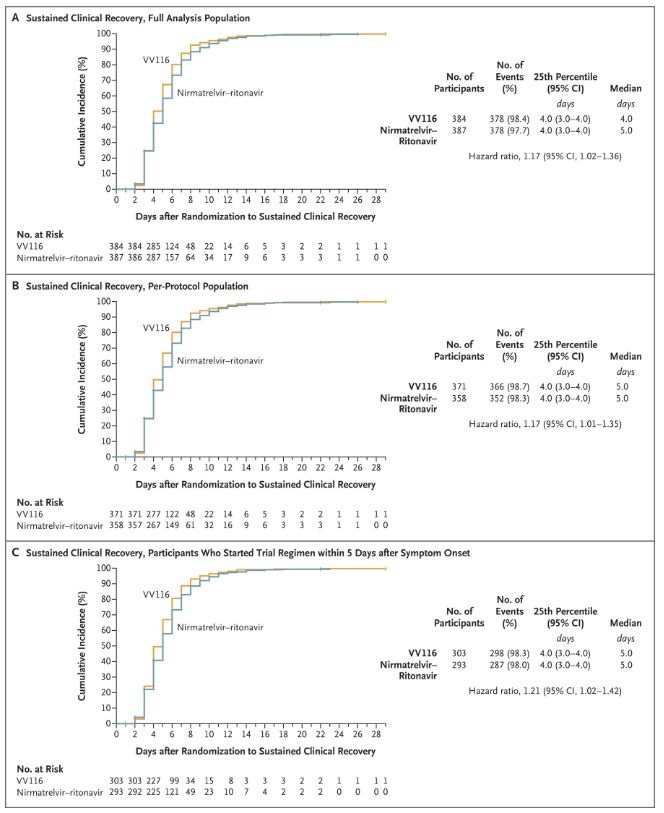
Mantener el tiempo de recuperación clínica
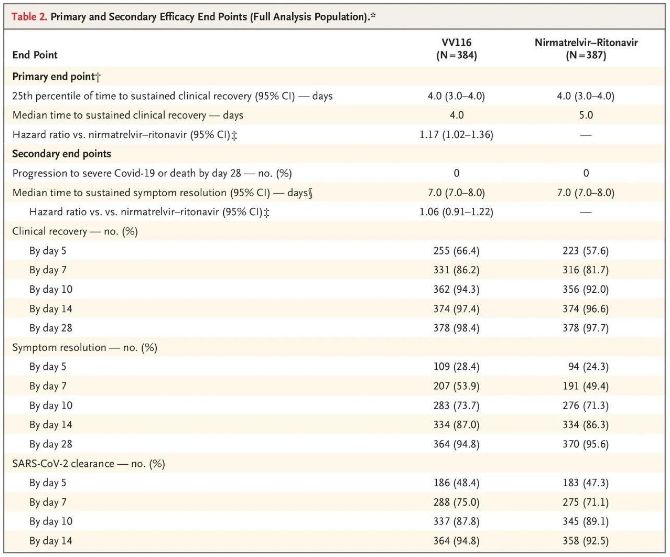
Criterios de valoración de eficacia primarios y secundarios (análisis exhaustivo de la población)
Fuente de la imagen: Referencia 2
En términos de seguridad, los participantes que recibieron VV116 informaron menos eventos adversos (67,4 %) que los que recibieron Paxlovid (77,3 %) en el seguimiento de 28 días, y la incidencia de eventos adversos de grado 3/4 fue menor para VV116 (2,6 %) que para Paxlovid (5,7 %).
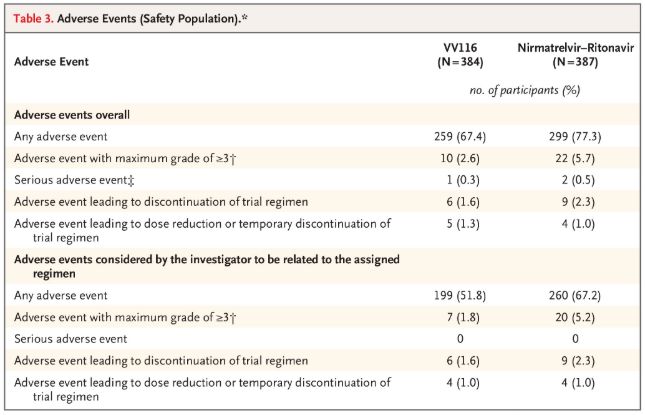
Eventos adversos (personas seguras)
Fuente de la imagen: Referencia 2
Controversias y preguntas
El 23 de mayo de 2022, Juniper anunció que el estudio clínico de registro de fase III de VV116 frente a PAXLOVID para el tratamiento temprano de la COVID-19 leve a moderada (NCT05341609) alcanzó su objetivo principal.

Fuente de la imagen: Referencia 1
En un momento en que faltaban detalles del ensayo, la controversia en torno al estudio de fase III era doble: en primer lugar, se trataba de un estudio simple ciego y, al no contar con un grupo de control con placebo, se temía que fuera difícil juzgar el fármaco de forma completamente objetiva; en segundo lugar, existían dudas sobre los criterios de valoración clínicos.
Los criterios de inclusión clínica para Juniper son: (i) resultados positivos en la prueba de la nueva corona, (ii) uno o más síntomas leves o moderados de COVID-19, y (iii) pacientes con alto riesgo de COVID-19 grave, incluyendo la muerte. Sin embargo, el único criterio de valoración clínico principal es el "tiempo hasta la recuperación clínica sostenida".
Justo antes del anuncio, el 14 de mayo, Juniper había revisado los criterios de valoración clínicos eliminando uno de los criterios de valoración clínicos primarios, “proporción de conversiones a enfermedad grave o muerte” [3].
![]()
Fuente de la imagen: Referencia 1
Estos dos puntos principales de controversia también se abordaron específicamente en el estudio publicado.
Debido al repentino brote de Omicron, la producción de comprimidos de placebo para Paxlovid no se había completado antes del inicio del ensayo y, por lo tanto, los investigadores no pudieron realizarlo con un diseño doble ciego y doble simulado. En cuanto al diseño simple ciego del ensayo clínico, Juniper indicó que el protocolo se elaboró tras comunicarse con las autoridades reguladoras y que este diseño implica que ni el investigador (incluido el evaluador del criterio de valoración del estudio) ni el patrocinador conocerán la asignación específica del fármaco terapéutico hasta que se cierre la base de datos final al término del estudio.
Hasta el momento del análisis final, ninguno de los participantes en el ensayo había experimentado muerte o progresión a un evento grave de Covid-19, por lo que no se pueden extraer conclusiones sobre la eficacia de VV116 para prevenir la progresión a Covid-19 grave o crítico o la muerte. Los datos indicaron que el tiempo medio estimado desde la aleatorización hasta la regresión sostenida de los síntomas objetivo relacionados con Covid-19 fue de 7 días (IC del 95 %, 7 a 8) en ambos grupos (cociente de riesgos, 1,06; IC del 95 %, 0,91 a 1,22) [2]. No es difícil explicar por qué se eliminó el criterio de valoración principal de "tasa de conversión a enfermedad grave o muerte", que se estableció originalmente antes del final del ensayo.
El 18 de mayo de 2022, la revista Emerging Microbes & Infections publicó los resultados del primer ensayo clínico de VV116 en pacientes infectados con la variante Omicron [4], un estudio de cohorte prospectivo y abierto con 136 pacientes hospitalizados confirmados.
Los datos del estudio mostraron que los pacientes con infección por Omicron que utilizaron VV116 dentro de los 5 días posteriores a su primera prueba de ácido nucleico positiva tuvieron un tiempo de regresión del ácido nucleico de 8,56 días, menor que los 11,13 días del grupo de control. La administración de VV116 a pacientes sintomáticos dentro del período de este estudio (2-10 días después de la primera prueba de ácido nucleico positiva) redujo el tiempo de regresión del ácido nucleico en todos los pacientes. En cuanto a la seguridad del fármaco, no se observaron efectos adversos graves en el grupo de tratamiento con VV116.
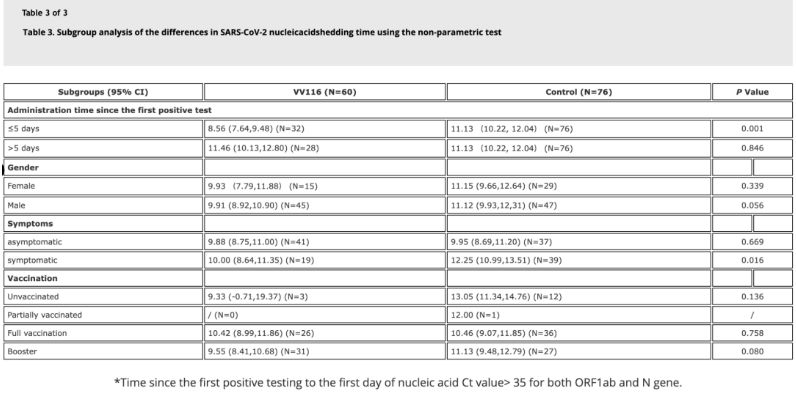
Fuente de la imagen: Referencia 4
Actualmente hay tres ensayos clínicos en curso sobre VV116, dos de los cuales son estudios de fase III para COVID-19 leve a moderado (NCT05242042, NCT05582629). El otro ensayo, para COVID-19 moderado a grave, es un estudio clínico internacional multicéntrico, aleatorizado y doble ciego de fase III (NCT05279235) para evaluar la eficacia y seguridad de VV116 en comparación con el tratamiento estándar. Según el anuncio de Juniper, el primer paciente fue reclutado y recibió la dosis en marzo de 2022.

Fuente de la imagen: clinicaltrials.gov
Referencias:
[1] Junshi Biotech: Anuncio sobre el criterio de valoración principal del estudio clínico registrado de fase III de VV116 frente a PAXLOVID para el tratamiento temprano de COVID-19 leve a moderado
[2]https://www.nejm.org/doi/full/10.1056/NEJMoa2208822?query=featured_home[3]https://clinicaltrials.gov/ct2/show/record/NCT05341609[4] Ensi Ma, Jingwen Ai, Yi Zhang, Jianming Zheng, Xiaogang Gao, Junming Xu, Hao Yin, Zhiren Fu, Hao Xing, Li Li, Liying Sun, Heyu Huang, Quanbao Zhang, Linlin Xu, Yanting Jin, Rui Chen, Guoyue Lv, Zhijun Zhu, Wenhong Zhang, Zhengxin Wang. (2022) Perfil de infecciones por Omicron y estado de vacunación entre 1881 receptores de trasplante de hígado: una cohorte retrospectiva multicéntrica. Microbios e infecciones emergentes 11:1, páginas 2636-2644.
Hora de publicación: 06-ene-2023
 Configuración de privacidad
Configuración de privacidad